
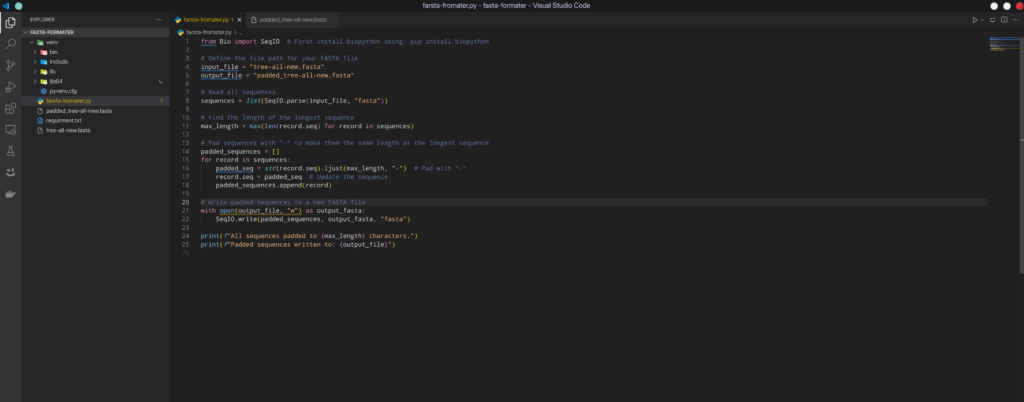
This Python script processes FASTA files to ensure that all sequences have the same length by padding shorter sequences with gaps ("-") and trimming longer sequences. It identifies the longest sequence in the file as the reference length and adjusts all other sequences accordingly. This normalization is crucial for bioinformatics analyses, such as phylogenetic tree construction and multiple sequence alignment, where input sequences must be uniform in length.
This Python script processes FASTA files to ensure that all sequences have the same length by padding shorter sequences with gaps ("-") and trimming longer sequences. It identifies the longest sequence in the file as the reference length and adjusts all other sequences accordingly. This normalization is crucial for bioinformatics analyses, such as phylogenetic tree construction and multiple sequence alignment, where input sequences must be uniform in length.
This script is useful in a variety of bioinformatics applications:
biopython for parsing and processing FASTA files
This script is ideal for bioinformaticians and researchers working with sequence data in evolutionary, molecular, and genomic studies. It ensures compatibility with tools like IQ-TREE and other alignment or phylogenetic software, making it an essential part of the bioinformatics data preprocessing pipeline.
This Python script processes FASTA files to ensure that all sequences have the same length by padding shorter sequences with gaps ("-") and trimming longer sequences. It identifies the longest sequence in the file as the reference length and adjusts all other sequences accordingly. This normalization is crucial for bioinformatics analyses, such as phylogenetic tree construction and multiple sequence alignment, where input sequences must be uniform in length.